

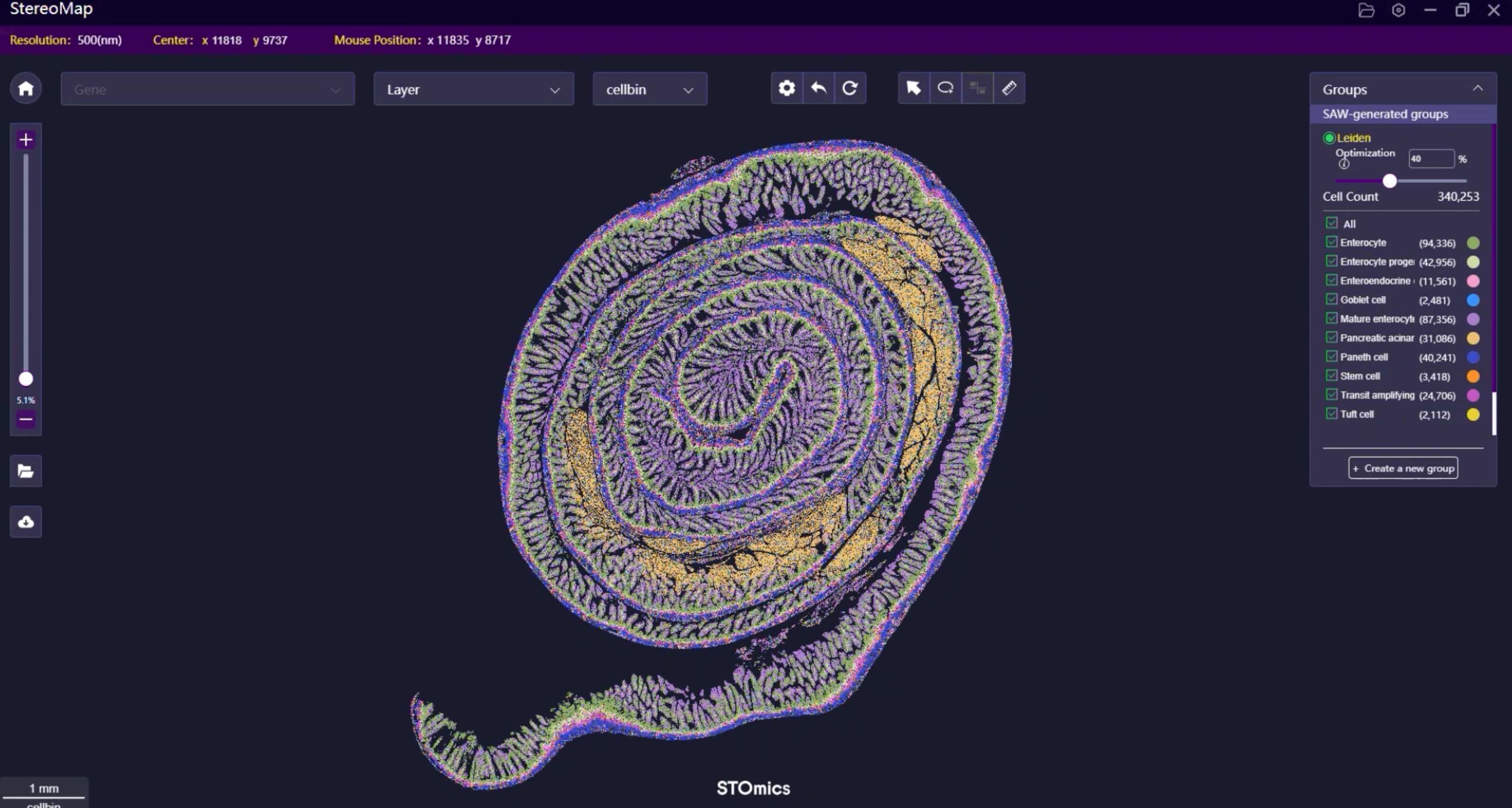
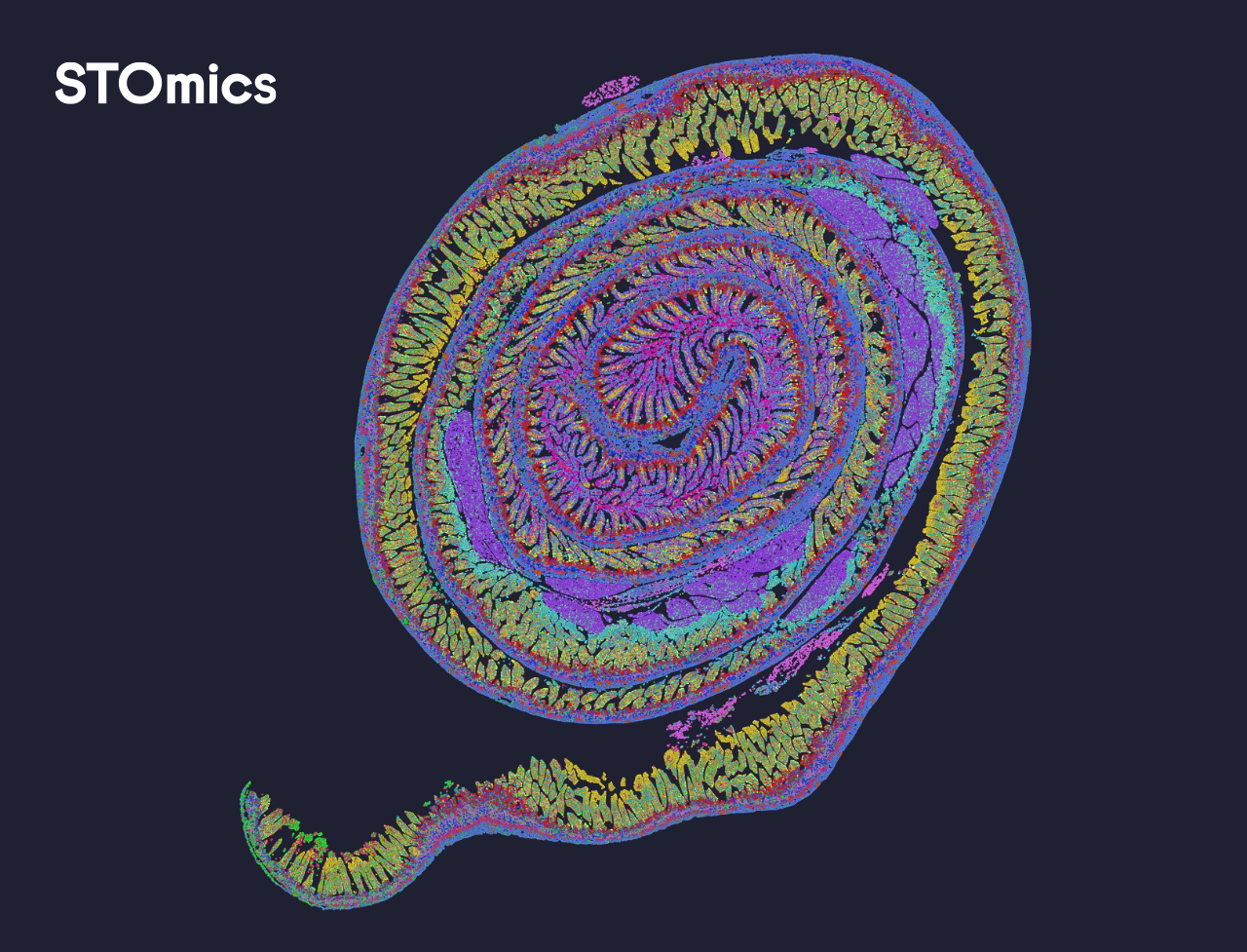
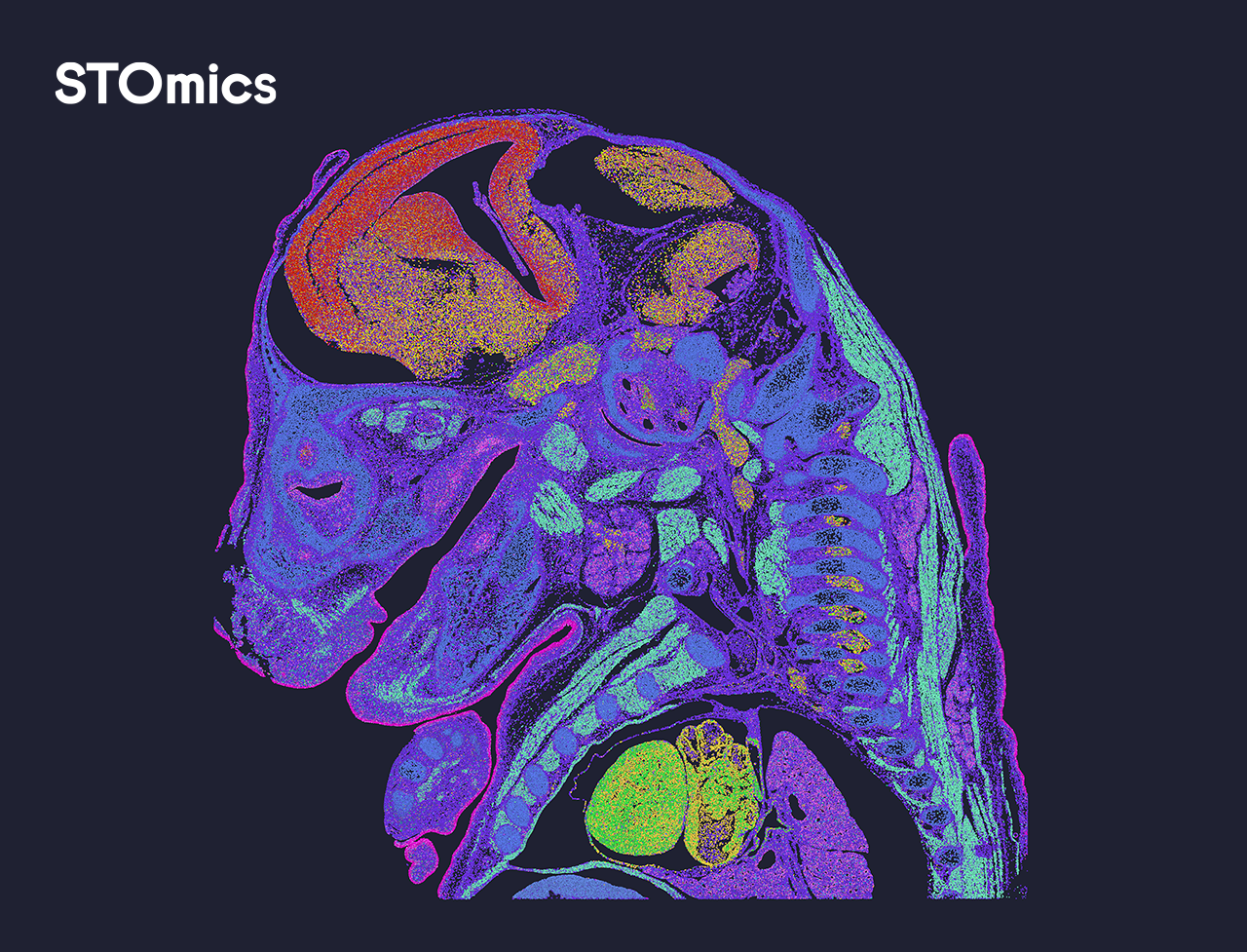

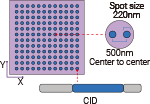





Yes, the DNB libraries with Stereo-seq OMNI FFPE V1.1 libraries and Stereo-seq FF V1.3 libraries can be pooled on the same FC for sequencing, and the Stereo-seq FF V1.3 libraries are not limited by the chip sizes.
All
Products
Resources
News
FAQ
Search