
All
Products
Resources
News
FAQ
Search


$ tree . |-- image | |-- SS200000135TL_D1_20220527_201353_1.1.0.ipr | `-- SS200000135TL_D1_20220527_201353_1.1.0.tar.gz |-- mask | `-- SS200000135TL_D1.barcodeToPos.h5 |-- md5 |-- reads | |-- E100026571_L01_trim_read_1.fq.gz | `-- E100026571_L01_trim_read_2.fq.gz `-- reference |-- STAR_SJ100 | |-- Genome | |-- SA | |-- SAindex | |-- chrLength.txt | |-- chrName.txt | |-- chrNameLength.txt | |-- chrStart.txt | |-- exonGeTrInfo.tab | |-- exonInfo.tab | |-- geneInfo.tab | |-- genomeParameters.txt | |-- genomeParameters_bkp.txt | |-- sjdbInfo.txt | |-- sjdbList.fromGTF.out.tab | |-- sjdbList.out.tab | `-- transcriptInfo.tab |-- genes.gtf `-- genome.fa 5 directories, 24 files
step | output | File Description |
|---|
splitMask | *.SN.barcodeToPos.bin | Split Stereo-seq Chip T mask file according to the CID. |
mapping | lane.Aligned.sortedByCoord.out.bam | Binary Alignment/Map file, used for storing the sequence alignment information. |
lane.barcodeReadsCount.txt | BMapped CID list file with reads counts for each CID, the three columns record x, y, and read count. | |
lane.Log.final.out | Summary mapping statistics after mapping job is complete (STAR output). | |
lane.Log.out | Main log file in STAR mapping (STAR output). | |
lane.Log.progress.out | Reports job process statistics (STAR output). | |
lane.SJ.out.tab | Splice junctions detected in the mapping (STAR output). | |
lane.bcPara | A parameters file defines CID mapping options. | |
lane_barcodeMap.stat | Statistical report of CID mapping, such as CID mapped reads count, reads sequencing quality, mapped DNB count, etc. | |
merge | SN.merge.barcodeReadsCount.txt | Mapped CID list file with reads counts for each CID., the three columns record x, y, and read count. |
count | SN.Aligned.sortedByCoord.out.merge. q10.dedup.target.bam | Annotated BAM file sorted by coordinate, includes uniquely mapped reads and multi-mapped reads whose HI:i tag is 1. |
SN.Aligned.sortedByCoord.out.merge. q10.dedup.target.bam.csi | Index file of annotated BAM. | |
SN.Aligned.sortedByCoord.out.merge. q10.dedup.target.bam.summary.stat | Statistical summary report file for count. Total reads in filter&deduplication metrics stand for the total alignment record count in BAM. Pass filter and total reads in annotation metrics are the same, they represent uniquely mapped reads that will be used for annotation, MID correction and quantification. | |
SN.raw.gef | Gene expression file in hdf5 format. This is the first raw matrix that includes the expression information over a complete chip region. It only includes geneExp group for the bin size of 1. The origin of expression matrix has been calibrated to (0,0), and the offset x and y has been record as minX and minY in the attribute of geneExp/expression dataset. | |
SN_raw_barcode_gene_exp.txt | A space-separate file records coordinate, gene, MID, and count information. Which is prepared to be a sampling file that performs sequence saturation. The 5 columns are y, x, geneIndex, MIDIndex, readCount. | |
register & imageTools ipr2img | date-hh-mm-ss.log | Log file. |
SN or other user specified name for the image folder used when input into imageQC/imageStudio | This subdirectory stores raw small image pieces in TIFF format. | |
SN_0000_0000_YYYY-MM-DD_hh-mm-ss-n.tif | Small image pieces in TIFF format. | |
fov_stitched_transformed.tif | The stitched panoramic image that has pre-registered with the track line pattern template. So that it will not need to further rotate a non-right angle or scale. | |
The stitched panoramic image from DAPI/IF layers that has pre-registered with the track line pattern template. So that it will not need to further rotate a non-right angle or scale. | ||
Track line cross point template for the registered DAPI/IF image. Used for assessing registration result. | ||
SN_ | IPR format image process record file which records basic image information gathered from ImageQC/ImageStudio. | |
Registered cell segmentation binary image file from DAPI/IF layers in TIFF format. | ||
Registered panoramic image file from DAPI/IF layers in TIFF format. | ||
SN.rpi | Registered panoramic image file, tissue area boundary, and cell boundaries (downsampled) that stores as an image pyramid. | |
SN_tissue_bbox.csv | Registered panoramic image size. | |
The tissue segmentation result of registered panoramic image file from DAPI/IF layers in TIFF format. | ||
Track line cross point template for the | ||
tissueCut | SN.gef | A gene expression file in hdf5 format. This file is a complete GEF format which includes geneExp group and wholeExp group in bin1, 10, 20, 50, 100, 200, and 500. It also includes a stat group. The origin of expression matrix has been calibrated to (0,0), and the offset x and y has been recorded as minX and minY in the attribute of geneExp/expression dataset. |
SN.tissue.gef | A gene expression file in hdf5 format. Tissue GEF includes the expression information of the tissue coverage region. It only includes geneExp group for the bin size of 1. The origin of expression matrix has been calibrated to (0,0), and the offset x and y has been recorded as minX and minY in the attribute of geneExp/expression dataset same to raw GEF. | |
SN.gem.gz | The compressed gene expression matrix that stores genes spatial expression data. | |
SN.tissue.gem.gz | The compressed gene expression matrix that stores genes spatial expression data in the tissue region. | |
tissue_fig | This directory stores the statistical plots for the tissue coverage region | |
scatter_100x100_MID_gene_counts.png | Scatter plot of MID count and gene number in each bin (bin100) | |
scatter_150x150_MID_gene_counts.png | Scatter plot of MID count and gene number in each bin (bin150) | |
scatter_200x200_MID_gene_counts.png | Scatter plot of MID count and gene number in each bin (bin200) | |
scatter_20x20_MID_gene_counts.png | Scatter plot of MID count and gene number in each bin (bin20) | |
scatter_50x50_MID_gene_counts.png | Scatter plot of MID count and gene number in each bin (bin50) | |
statistic_100x100_MID_gene_DNB.png | Univariate distribution of MID count, gene numbers, and DNB numbers with rug along the x axis (bin100) | |
statistic_150x150_MID_gene_DNB.png | Univariate distribution of MID count, gene numbers, and DNB numbers with rug along the x axis (bin150) | |
statistic_200x200_MID_gene_DNB.png | Univariate distribution of MID count, gene numbers, and DNB numbers with rug along the x axis (bin200) | |
statistic_20x20_MID_gene_DNB.png | Univariate distribution of MID count, gene numbers, and DNB numbers with rug along the x axis (bin20) | |
statistic_50x50_MID_gene_DNB.png | Univariate distribution of MID count, gene numbers, and DNB numbers with rug along the x axis (bin50) | |
violin_100x100_MID_gene.png | Violin plots show the distribution of deduplicated MID count and gene number in each bin (bin100) | |
violin_150x150_MID_gene.png | Violin plots show the distribution of deduplicated MID count and gene number in each bin (bin150) | |
violin_200x200_MID_gene.png | Violin plots show the distribution of deduplicated MID count and gene number in each bin (bin200) | |
violin_20x20_MID_gene.png | Violin plots show the distribution of deduplicated MID count and gene number in each bin (bin20) | |
violin_50x50_MID_gene.png | Violin plots show the distribution of deduplicated MID count and gene number in each bin (bin50) | |
tissuecut.stat | Statistical report for tissue coverage region. | |
cellCut | SN.cellbin.gef | A gene expression file for cells in hdf5 format. Cellbin GEF includes the expression information of the cells, such as the coordinate of the centroid, boundary coordinates, expression of genes, and cell area. The cell is record by its boundary. The origin of expression matrix has been calibrated to (0,0), and the coordinate offset x and y has been record as offsetX and offsetY in the attribute of the GEF file same to minX and minY in the raw GEF file. |
spatialCluster | SN.spatial.cluster.h5ad | H5AD file for spatial clustering analysis result. |
cellCluster | SN.cell.cluster.h5ad | H5AD file for cell clustering analysis result. |
saturation | plot_1x1_saturation.png | Sequencing saturation analysis plots for bin1. Calculated by 1- (unique reads/total reads) for each bin (bin1). |
plot_200x200_saturation.png | Sequencing saturation analysis plots for bin200. Calculated by 1- (unique reads/total reads) for each bin (bin1). | |
sequence_saturation.tsv | Sequence saturation file. The nine colums are sampling component (#sample), bin1 total reads (bar_x), sequence saturation value at bin1 (bar_y1), median gene count at bin1 (bar_y2), unique reads at bin1 (bar_umi), bin200 total reads (bin_x), sequence saturation value at bin200 (bin_y1), median gene count at bin200 (bin_y2), and unique reads at bin200 (bin_umi), respectively. | |
report | SN.report.html | HTML analytical report. |
SN.statistics.json | Statistical summary report in JSON format. It gathers all important statistical metrics from the statistical report in each step. | |
scatter_1x1_MID_gene_counts.png | Scatter plot of MID count and gene number in each cell | |
statistic_1x1_MID_gene_DNB.png | Univariate distribution of MID count, gene numbers, and DNB numbers with rug along the x axis of each cell | |
violin_1x1_MID_gene.png | Violin plots show the distribution of deduplicated MID count and gene number in each cell |
terminate called after throwing an instance of 'std::invalid_argument' what(): Could not open the file: xxxxxx ...
#006: H5FDsec2.c line 352 in H5FD__sec2_open(): unable to open file: name = '/path/to/hdf5/file', errno = 2, error message = 'No such file or directory', flags = 0, o_flags = 0 major: File accessibility minor: Unable to open file ...
# Attempt 1: Bind file path before execute command. $ export SINGULARITY_BIND="/path/to/file/directory" # Attempt 2: Turn off the HDF5 lock on the file by running the command below before running SAW. $ export HDF5_USE_FILE_LOCKING=FALSE
EXITING: FATAL INPUT ERROR: empty value for parameter “readNameSeparator” in input “Command-Line” SOLUTION: use non-empty value for this parameter
Error code: SAW-A10183 EXITING because of fatal ERROR: not enough memory for BAM sorting: SOLUTION: re-run STAR with at least –limitBAMsortRAM xxxxxxxxx
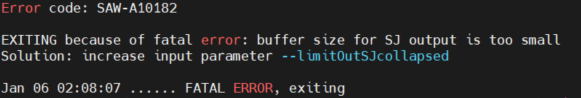
EXITING because of fatal error: buffer size of SJ output is too small Solution: increase input parameter --limitOutSJcollapsed
OSError: [Errno 30] Read-only file system: '/opt/saw_v5.4.0_software/pipeline/imageTools/imagetools-1.0.0/log'
Pipeline | Code | Error type | Examples and error handling |
|---|
splitMask | SAW-A00001 | Parameters invalid or missing | e.g. "parameters error"Please check your input parameters. Some required parameters might be missed. e.g. "splitBcPos error, expected 1_24 or 2_25" Please check your input CID position is either 1_24 or 2_25. |
SAW-A00002 | File open failed | Please check the input file exists and has the correct access permission. | |
SAW-A00003 | File parse failed | e.g. "only support .bin or .h5 file." Please check your input file is in the correct file format. | |
SAW-A00004 | File open failed | e.g. "cannot write to file, /path/to/file" Please check your output directory path is an existing path. | |
SAW-A00005 | Other API error | Please check Appendix C or contact FAS/FBS for help. | |
SAW-A00006 | Software exception | Please check Appendix C or contact FAS/FBS for help. | |
CIDCount | SAW-A00021 | Parameters invalid or missing | Please check your input parameters. Some required parameters might be missed. |
SAW-A00022 | File open failed | e.g. "cannot open such file, /path/to/file" Please check the input file exists and has the correct access permission. | |
SAW-A00023 | Failed to parser the file | Please check your input file is in the correct file format. | |
SAW-A00024 | Other API error | Please check Appendix C or contact FAS/FBS for help. | |
SAW-A00025 | Software exception | Please check Appendix C or contact FAS/FBS for help. | |
mapping | SAW-A10101 | Parameters invalid or missing | e.g. "EXITING: FATAL INPUT ERROR: unrecognized parameter name "outSAMattribute" in input "Command-Line-Initial"" Please check the spelling of your argument and parameters. e.g. "please check the umi position and length" Please check the length of the reads in FQ1 are consistent with the parameters set for barcode length and umi length. For example, the length of the sum of barcode and umi set in bcPara file is 35 bp, but one of the reads in FQ1 is 30 bp, then you will see A10101 error code. Please check the parameters set in the bcPara file are consistent with your FQs. |
SAW-A10102 | File open failed | e.g. "Error, cannot open the file which be expected in gz or ascii format" Please check the file permission and file format. e.g. "barcodePositionMapFile does not exists: /path/to/mask" Please check the mask file exists and has the correct access permission. | |
SAW-A10103 | File parse failed | e.g. "sequence and quality have different length" Please check the completeness of the reads in FQ. This issue may arise if the FQs are in incorrect format or the file was incompletely written or transferred. | |
SAW-A10104 | Invalid data or data exception | Error data. Please check the file format and content. | |
SAW-A10105 | File deletion failed | This error arose if "_STARtmp" directory failed to be deleted. Please check whether the program has completely finished according to the *.Log.progress.out or *_barcodeMap.stat file. | |
SAW-A10106 | File IO failed | Please contact FAS/FBS for help. | |
SAW-A10107 | Failure on APIs of system and libraries | Please contact FAS/FBS for help. | |
SAW-A10108 | Software assert | Please contact FAS/FBS for help. | |
SAW-A10109 | Software exception | Please contact FAS/FBS for help. | |
SAW-A10110 | Allocate memory error | This error occurs when you run out of RAM. You may need to kill some jobs that use the same RAM or upgrade your hardware to get more RAM resources for your job. | |
SAW-A10111 | Out of disk space | This error occurs when you store too many files on your hard disk. Please remove some files to free disk space. | |
SAW-A10200 | Parameter missing | Please check your input parameters. Some required parameters might be missed. | |
SAW-A10201 | CID comparison rate is too low | This error usually arises because the CID information of the input FQs is not the same as the CID in the input mask file. Please use the correct SN-FQ pairs for mapping. | |
SAW-A10202 | Fail to create the index for the BAM file | Please contact FAS/FBS for help. | |
SAW-A10300 | Fail to load indexed reference | Please check the existence, access permission, and completeness for the indexed reference. | |
count | SAW-A20001 | Parameter missing | Please check your input parameters. Some required parameters might be missed. |
SAW-A20002 | File open failed | Please check the input file exists and has the correct access permission. | |
SAW-A20003 | File parse failed | Please check your input BAM header is in the correct file format. | |
SAW-A20004 | Allocate memory error | This error occurs when you run out of RAM. You may need to kill some jobs that use the same RAM or upgrade your hardware to get more RAM resources for your job. | |
SAW-A20009 | Software exception | Please contact FAS/FBS for help. | |
SAW-A20101 | Parameter missing | Please check your input parameters. Some required parameters might be missed. | |
SAW-A20102 | File open failed | Please check the file that exists and has the correct access permission. | |
SAW-A20103 | File parse failed | Please check your input file has a valid column number. | |
merge | SAW-A30001 | Parameter missing | Please check your input parameters. Some required parameters might be missed. |
SAW-A30002 | File open failed | Please check the input file exists and has the correct access permission. | |
SAW-A30003 | File parse failed | Please check your mask file is in the correct file format. Only support .h5/.bin mask file. | |
SAW-A30004 | Fail to create output file | Fail to create output file. Please check your writing permission of the output directory. | |
SAW-A30005 | Fail to open input file | Fail to open input TXT file. Please check the file that exists and have the correct access permission. | |
SAW-A30006 | Out of index | Record coordinates exceed the expected range. Please check the input file is the output of mapping. | |
SAW-A30007 | Allocate memory error | Please check whether the range of the coordinates is too large, or you have run out of RAM. You may need to kill some jobs that use the same RAM or upgrade your hardware to get more RAM resources for your job. | |
register & rapidRegister | SAW-A40001 | Parameter missing | Please check your input parameters. Some required parameters might be missed. |
SAW-A40002 | File open failed | Please check the input file that exists and has the correct access permission. Or, please check whether a stitched panoramic TIFF (.tif or .tiff) image exists in the TAR.GZ. | |
SAW-A40003 | File parse failed | Please check your input file is in the correct file format. The -v input gene expression matrix should be either a *tsv, barcode_gene_exp.txt, *.gem.gz, or *raw.gef. | |
SAW-A40004 | Invalid data or data exception | Error data. Please check the file content. This error may arise because the -v input file is empty, the CZI file in TAR.GZ is invalid, or the QCPassFlag in IPR is 0. | |
SAW-A40005 | Tissue segmentation error | Abnormal tissue segmentation reference score in image preprocessing. | |
SAW-A40006 | IPR field missing | "Stitch/BGIStitch/StitchedGlobalLoc", does not exist. | |
SAW-A40007 | Tiled image missing | Please check the uncompressed image folder has tiled images. | |
imageTools | SAW-A40401 | Parameter missing | Please check your imageTools merge input parameters. Some required parameters might be missed. |
SAW-A40402 | File open failed | Please check the imageTools merge input file that exists and has the correct access permission. | |
SAW-A40405 | Invalid input | imageTools merge inputs of less than two images or more than three images. | |
SAW-A40406 | File pairing failed | Please check the imageTools merge input TIFF sizes are the same. Since the merge function is used for evaluating segmentation results, the input images are supposed to be the same in size and position (tissue position in the whole image). | |
SAW-A40501 | Parameter missing | Please check your imageTools overlay input parameters. Some required parameters might be missed. | |
SAW-A40502 | File open failed | Please check the imageTools overlay input file that exists and has the correct access permission. | |
SAW-A40504 | Invalid data or data exception | Error data. Please check the file content. This error may arise because the -c input IPR file does not include Stitch/TransformTemplate or Register/MatrixTemplate information | |
SAW-A40601 | Parameter missing | Please check your imageTools img2rpi input parameters. Some required parameters might be missed. | |
SAW-A40602 | File open failed | Please check the imageTools img2rpi input file that exists and has the correct access permission. | |
SAW-A40605 | Invalid input | imageTools img2rpi input -i and -g have different length. These two inputs are supposed to be paired. | |
SAW-A40701 | Parameter missing | Please check your imageTools ipr2img input parameters. Some required parameters might be missed. | |
SAW-A40702 | File open failed | Please check the imageTools ipr2imginput file exists and has the correct access permission. Or, please check whether a stitched panoramic TIFF (.tif or .tiff) image exists in the TAR.GZ. | |
SAW-A40703 | File parse failed | Please check your imageTools ipr2img input file is in the imageQC/imageStudio output TAR.GZ format. | |
SAW-A40704 | Invalid data or data exception | Error data. Please check the imageTools ipr2img input IPR file content. This error may arise because the CZI file in TAR.GZ is invalid, or the image has not either automatically or manually registered with the expression matrix. The second circumstance can be confirmed from IPR by checking whether the StereoResepSwitch/register is TRUE (has not performed automatic registration) or the ManualState/register is FALSE (has not performed manual registration). The third possible reason is the StereoResepSwitch/tissueseg is TRUE, therefore cannot output cell segmentation result. | |
SAW-A40706 | File pairing failed | Please check the shape of registered tissue segmentation images stored in the imageTools ipr2img input IPR are the same. Or please contact FAS/FBS for help. | |
tissueCut | SAW-A50001 | Parameter missing | Please check your input parameters. Some required parameters might be missed |
SAW-A50002 | File open failed | Please check the file that exists and has the correct access permission. | |
SAW-A50003 | File parse failed | Please check your input file is in the correct file format. | |
SAW-A50004 | Fail to create output file | Fail to create output file. Please check your writing permission of the output directory. | |
SAW-A50005 | Fail to write TIFF | Fail to write a TIFF file. | |
SAW-A50006 | h5AttrWrite error | Please check the H5 file attributes. | |
SAW-A50007 | h5DatasetWrite error | Please check the H5 file dataset. | |
cellCut | SAW-A60001 | Parameter missing | Please check your input parameters. Some required parameters might be missed |
SAW-A60002 | File open failed | Please check the file that exists and has the correct access permission. | |
SAW-A60003 | File parse failed | The file does not contain correct information. Please check the file format. | |
SAW-A60110 | Program version error | Please check your output GEF version. Your input GEF version might be too old. | |
SAW-A60111 | Call process error | e.g. "Please call freeRestriction first, or call restrictRegion function before restrictGene." This error arose because the invocation flow order was messed up. Please modify your invocation flow as prompted. | |
SAW-A60120 | Invalid data or data exception | Error data. Please check the file content. | |
SAW-A60121 | File information missing | Failed to read the file. Please check whether the file is damaged. | |
SAW-A60122 | File pairing failed | Please check the TIFF mask size is consistent with the size of expression matrix. Since the mask has been registered with the expression matrix, their sizes are supposed to be the same. | |
SAW-A60130 | Fail to create output file | Fail to create output H5 file. Please check your writing permission of the output directory or contact FAS/FBS for help. | |
SAW-A60140 | Allocate memory error | This error occurs when you run out of RAM. You may need to kill some jobs that use the same RAM or upgrade your hardware to get more RAM resources for your job. | |
SAW-A60150 | Dimensions of gene expression matrix did not match | Please contact FAS/FBS for help. | |
spatialCluster | SAW-A70001 | Parameter missing | e.g. "-i or --gef_file is missing" Please check your input parameters. Some required parameters might be missed. |
SAW-A70002 | File open failed | e.g. “cannot access /path/to/file: No such file or directory.” Please check the file that exists and has the correct access permission. | |
SAW-A70005 | Value error | e.g. "The bin size is out of range, please check the range of gef binsize is in [1,10,20,50,100,200,500]." Please reset the bin size as prompted. e.g. "Gene number less than 3000, please check your gef file" Please check the content of your GEF file, and make sure there are at least 3000 genes for clustering. | |
cellCluster | SAW-A70101 | Parameter missing | e.g. "-i or --gef_file is missing" Please check your input parameters. Some required parameters might be missed. |
SAW-A70102 | File open failed | e.g. “cannot access /path/to/file: No such file or directory.” Please check the file that exists and has the correct access permission. | |
SAW-A70105 | Value error | e.g. "The bin size is out of range, please check the range of gef binsize is in [1,10,20,50,100,200,500]." Please reset the bin size as prompted. e.g. "Gene number less than 3000, please check your gef file" Please check the content of your GEF file, and make sure there are at least 3000 genes for clustering. | |
saturation | SAW-A80001 | Parameter missing | e.g. "-i is missing." Please check your input parameters. Some required parameters might be missed. |
SAW-A80002 | File open failed | Please check the file that exists and has the correct access permission. | |
SAW-A80003 | File parse failed | Invalid GEF file. Please check the file format. | |
SAW-A80004 | Invalid data or data exception | e.g. "no data left after filter by coordinates." Please check the file content. | |
SAW-A80005 | Invalid data or data exception | e.g. "total map reads is 0, please check file format from --bcstat" Please check the file content of the input file as prompted. | |
SAW-A80006 | File pairing failed | e.g. "map reads less than annotated reads." Please check the input mapping statistical report and the count statistical report are from the same analysis. | |
SAW-A80007 | Plot error | Please contact FAS/FBS for help. PATH environment may not have python3. | |
SAW-A80008 | Fail to create output file | Fail to generate saturation file, please contact FAS/FBS for help. | |
report | SAW-A90001 | Parameter missing | e.g. "-m or --barcodeMapStat is missing." Please check your input parameter as prompted. Some required parameters might be missed. |
SAW-A90002 | File open failed | e.g. "cannot access *: No such file or directory." Please check the file that exists and has the correct access permission. | |
SAW-A90003 | File parse failed | JSON file format error. This error may arise because the input statistics files were not generated in the same SAW version as report. Or, the input mapping file prefix can not be parsed. Please contact FAS/FBS for help. | |
SAW-A90004 | Invalid data or data exception | e.g."information loss: fail to find 'bin_[size]' or 'ssDNA' in '*.rpi'." Please check the file content. | |
SAW-A90005 | Fail to create output file | Fail to create output file. Please check your writing permission of the output directory. | |
manualRegister | SAW-A40801 | Parameter missing | Please check your input parameters. Some required parameters might be missed |
SAW-A40802 | File open failed | Please check the file that exists and has the correct access permission. Or, please check whether the pre-registered image fov_stitched_transformed.tif exists in the input directory. | |
SAW-A40803 | File parse failed | Please check your input file is in the correct file format. The -v input gene expression matrix should be either a *tsv, barcode_gene_exp.txt, *.gem.gz, or *raw.gef. | |
SAW-A40804 | Invalid data or data exception | Error data. Please check the file content. This error may arise because the -v input file is empty. The second possible reason is that the gene expression matrix information in the IPR Register module (MatrixShape, Xstart, Ystart) does not match with the input GEF file (minX, minY, maxX, maxY), because the manual registration has to be processed on the identical matrix. |